



RT-qPCR ត្រូវបានបង្កើតឡើងពីបច្ចេកវិទ្យា PCR ធម្មតា។វាបន្ថែមសារធាតុគីមី fluorescent (ថ្នាំពណ៌ fluorescent ឬ fluorescent probes) ទៅក្នុងប្រព័ន្ធប្រតិកម្ម PCR ប្រពៃណី និងរកឃើញដំណើរការបិទភ្ជាប់ PCR និងដំណើរការបន្ថែមក្នុងពេលវេលាជាក់ស្តែង យោងទៅតាមយន្តការ luminescent ផ្សេងគ្នារបស់ពួកគេ។ការផ្លាស់ប្តូរសញ្ញា fluorescent នៅក្នុងឧបករណ៍ផ្ទុកត្រូវបានប្រើដើម្បីគណនាបរិមាណនៃការផ្លាស់ប្តូរផលិតផលនៅក្នុងវដ្តនីមួយៗនៃ PCR ។បច្ចុប្បន្ននេះវិធីសាស្រ្តទូទៅបំផុតគឺវិធីសាស្ត្រជ្រលក់ពណ៌ fluorescent និងវិធីសាស្ត្រស៊ើបអង្កេត។
វិធីសាស្រ្តនៃការជ្រលក់ពណ៌ fluorescent៖
សារធាតុពណ៌ fluorescent មួយចំនួនដូចជា SYBR Green Ⅰ, PicoGreen, BEBO ជាដើម មិនបញ្ចេញពន្លឺដោយខ្លួនឯងទេ ប៉ុន្តែបញ្ចេញពន្លឺបន្ទាប់ពីភ្ជាប់ទៅនឹងចង្អូរតូចៗនៃ dsDNA ។ដូច្នេះនៅដើមដំបូងនៃប្រតិកម្ម PCR ម៉ាស៊ីនមិនអាចរកឃើញសញ្ញា fluorescent បានទេ។នៅពេលដែលប្រតិកម្មបន្តទៅផ្នែកបន្ថែមការបន្ថែម (វិធីសាស្ត្រពីរជំហាន) ឬដំណាក់កាលបន្ថែម (វិធីសាស្ត្របីជំហាន) ខ្សែពីរត្រូវបានបើកនៅពេលនេះ ហើយ DNA polymerase ថ្មី កំឡុងពេលសំយោគខ្សែ ម៉ូលេគុល fluorescent ត្រូវបានបញ្ចូលគ្នានៅក្នុងចង្អូរតូច dsDNA និងបញ្ចេញ fluorescence ។នៅពេលដែលចំនួននៃវដ្ត PCR កើនឡើង ការជ្រលក់ពណ៌កាន់តែច្រើនបញ្ចូលគ្នាជាមួយ dsDNA ហើយសញ្ញា fluorescent ក៏ត្រូវបានពង្រឹងជាបន្តបន្ទាប់ផងដែរ។យក SYBR Green Ⅰ ជាឧទាហរណ៍។
វិធីសាស្ត្រស៊ើបអង្កេត៖
ការស៊ើបអង្កេត Taqman គឺជាឧបករណ៍ស្ទង់អ៊ីដ្រូលីស៊ីសដែលប្រើជាទូទៅបំផុត។មានក្រុម fluorescent នៅចុង 5′ នៃការស៊ើបអង្កេត ជាធម្មតា FAM ។ការស៊ើបអង្កេតខ្លួនវាគឺជាលំដាប់ដែលបំពេញបន្ថែមទៅនឹងហ្សែនគោលដៅ។មានក្រុម fluorescent quenching នៅចុង 3′ នៃ fluorophore ។យោងទៅតាមគោលការណ៍នៃការផ្ទេរថាមពល fluorescent resonance (Förster resonance energy transfer, FRET) នៅពេលដែលក្រុម fluorescent អ្នកយកព័ត៌មាន (ម៉ូលេគុល fluorescent ម្ចាស់ជំនួយ) និងក្រុម fluorescent quenching (acceptor fluorescent molecule) នៅពេលដែលវិសាលគមរំភើបត្រួតលើគ្នា និងចំងាយគឺនៅជិតគ្នាខ្លាំង (7-10nm) នៃ fluorescent ទទួលយក ម៉ូលេគុលរបស់ fluorescent ។ ម៉ូលេគុលខណៈពេលដែល autofluorescence ត្រូវបានចុះខ្សោយ។ដូច្នេះហើយ នៅដើមដំបូងនៃប្រតិកម្ម PCR នៅពេលដែលការស៊ើបអង្កេតគឺឥតគិតថ្លៃ និងនៅដដែលនៅក្នុងប្រព័ន្ធ ក្រុម fluorescent អ្នករាយការណ៍នឹងមិនបញ្ចេញ fluorescence ទេ។នៅពេលដែល annealing, primer និង probe ចងទៅនឹងគំរូ។ក្នុងកំឡុងដំណាក់កាលពង្រីក សារធាតុប៉ូលីមេរ៉ាសបន្តសំយោគខ្សែសង្វាក់ថ្មី។DNA polymerase មានសកម្មភាព 5'-3' exonuclease ។នៅពេលឈានដល់ការស៊ើបអង្កេត DNA polymerase នឹង hydrolyze ការស៊ើបអង្កេតពីគំរូ បំបែកក្រុម fluorescent អ្នករាយការណ៍ពីក្រុម quencher fluorescent និងបញ្ចេញសញ្ញា fluorescent ។ដោយសារមានទំនាក់ទំនងមួយទល់នឹងមួយរវាងការស៊ើបអង្កេត និងគំរូ វិធីសាស្ត្រស៊ើបអង្កេតគឺប្រសើរជាងវិធីសាស្ត្រជ្រលក់ក្នុងលក្ខខណ្ឌនៃភាពត្រឹមត្រូវ និងភាពប្រែប្រួលនៃការធ្វើតេស្ត។
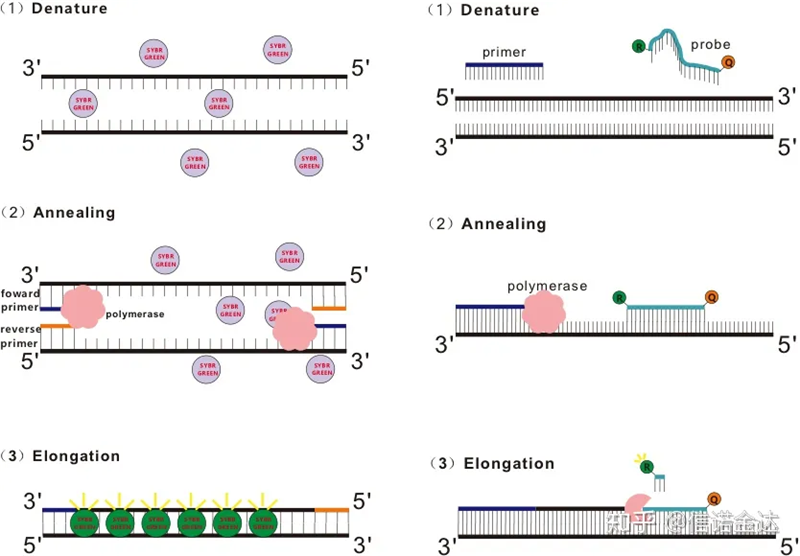
រូបភាពទី 1 គោលការណ៍នៃ qRT-PCR
ការរចនាបឋម
គោលការណ៍៖
ថ្នាំ primers គួរតែត្រូវបានរចនាឡើងនៅក្នុងតំបន់អភិរក្សនៃស៊េរីអាស៊ីត nucleic និងមានភាពជាក់លាក់។
វាជាការល្អបំផុតក្នុងការប្រើលំដាប់ cDNA ហើយលំដាប់ mRNA ក៏អាចទទួលយកបានផងដែរ។បើមិនដូច្នោះទេ សូមស្វែងរកការរចនាតំបន់ cds នៃលំដាប់ DNA។
ប្រវែងនៃផលិតផលបរិមាណ fluorescent គឺ 80-150bp វែងបំផុតគឺ 300bp ប្រវែង primer ជាទូទៅនៅចន្លោះ 17-25 bases ហើយភាពខុសគ្នារវាង primers ខាងលើ និងខាងក្រោមមិនគួរធំពេកទេ។
មាតិកា G + C ស្ថិតនៅចន្លោះពី 40% ទៅ 60% ហើយ 45-55% គឺល្អបំផុត។
តម្លៃ TM ស្ថិតនៅចន្លោះ 58-62 ដឺក្រេ។
ព្យាយាមជៀសវាង primer dimers និង self-dimers (កុំបង្ហាញលើសពី 4 គូនៃមូលដ្ឋានបន្ថែមជាប់គ្នា) រចនាសម្ព័ន្ធ hairpin ប្រសិនបើមិនអាចជៀសបាន ធ្វើ ΔG<4.5kJ/mol* ប្រសិនបើអ្នកមិនអាចធានាថា gDNA ត្រូវបានដកចេញកំឡុងពេលចម្លងបញ្ច្រាស់ស្អាតទេ វាជាការល្អបំផុតក្នុងការរចនា primers នៃ intron * 3′ បន្ត និងមិនអាចកែប្រែបាន រចនាសម្ព័ន្ធ G/C សំបូរបែប ជៀសវាងតំបន់ G/C ។ 2-3) primers និង non-
ភាពជាក់លាក់ ភាពដូចគ្នានៃលំដាប់ដែលពង្រីកច្រើនដងគឺតិចជាង 70% ឬមាន 8 ភាពដូចគ្នានៃមូលដ្ឋាន។
មូលដ្ឋានទិន្នន័យ៖
ស្វែងរក CottonFGD តាមពាក្យគន្លឹះ
ការរចនាបឋម៖
ការរចនាបឋម IDT-qPCR
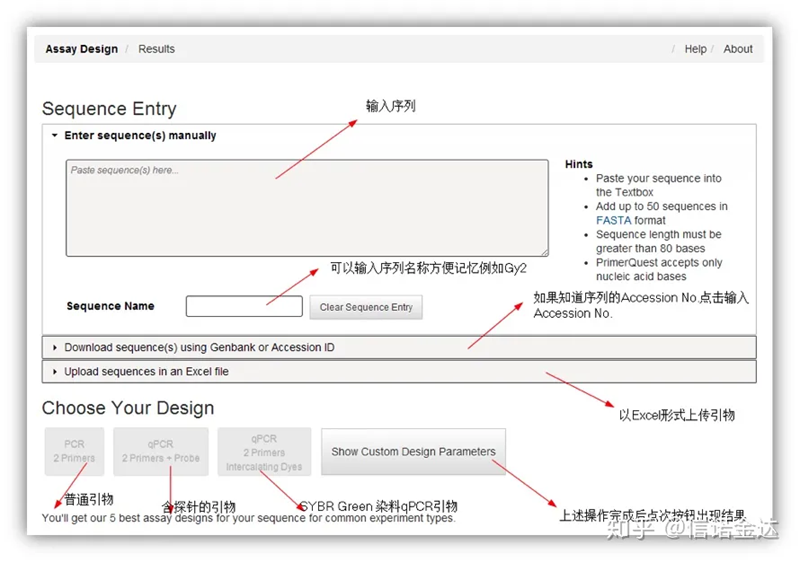
Fig2 IDT ទំព័រឧបករណ៍រចនាបឋមតាមអ៊ីនធឺណិត
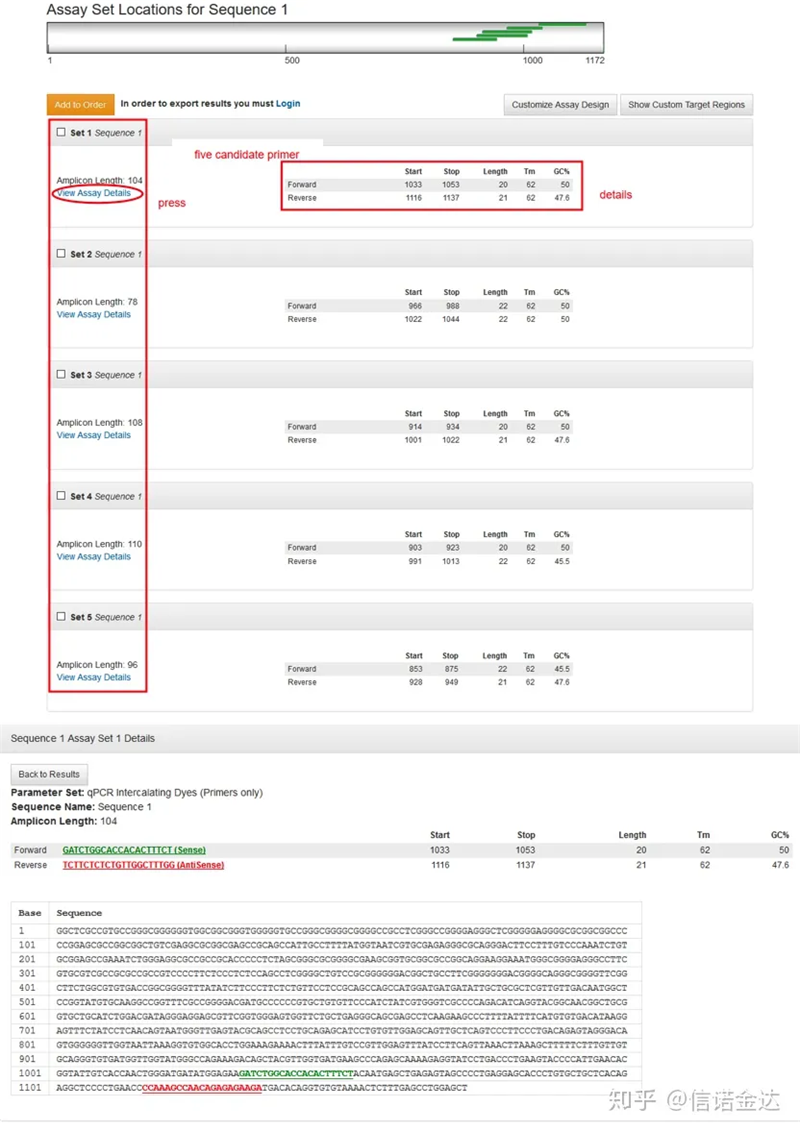
រូបភាពទី 3 បង្ហាញទំព័រលទ្ធផល
ការរចនានៃ lncRNA primers:
lncRNA៖ជំហានដូចគ្នានឹង mRNA ។
miRNA៖គោលការណ៍នៃវិធីសាស្រ្ត stem-loop: ដោយសារ miRNAs ទាំងអស់គឺជាស៊េរីខ្លីប្រហែល 23 nt ការរកឃើញ PCR ផ្ទាល់មិនអាចត្រូវបានអនុវត្តទេ ដូច្នេះឧបករណ៍លំដាប់លំដោយ stem-loop ត្រូវបានប្រើ។លំដាប់លំដោយ stem-loop គឺជា DNA ខ្សែតែមួយដែលមានប្រហែល 50 nt ដែលអាចបង្កើតរចនាសម្ព័ន្ធ hairpin ដោយខ្លួនវាផ្ទាល់។3 'ចុងបញ្ចប់អាចត្រូវបានរចនាឡើងជាលំដាប់ដែលបំពេញបន្ថែមទៅនឹងបំណែកផ្នែក miRNA បន្ទាប់មក miRNA គោលដៅអាចត្រូវបានតភ្ជាប់ទៅ stem-loop sequence កំឡុងពេលចម្លងបញ្ច្រាស ហើយប្រវែងសរុបអាចឈានដល់ 70bp ដែលស្របតាមប្រវែងនៃផលិតផលពង្រីកដែលកំណត់ដោយ qPCR ។ការរចនាបឋមនៃកន្ទុយ miRNA ។
ការរកឃើញជាក់លាក់នៃការពង្រីក៖
មូលដ្ឋានទិន្នន័យបំផ្ទុះតាមអ៊ីនធឺណិត៖ បំផ្ទុះ CottonFGD ដោយភាពស្រដៀងគ្នាលំដាប់
Local blast៖ យោងទៅលើការប្រើ Blast+ ដើម្បីធ្វើការផ្ទុះមូលដ្ឋាន linux និង macos អាចបង្កើតមូលដ្ឋានទិន្នន័យដោយផ្ទាល់ ប្រព័ន្ធ win10 ក៏អាចត្រូវបានធ្វើបន្ទាប់ពីដំឡើង ubuntu bash។បង្កើតមូលដ្ឋានទិន្នន័យបំផ្ទុះក្នុងស្រុក និងបំផ្ទុះមូលដ្ឋាន ;បើក ubuntu bash នៅលើ win10 ។
សេចក្តីជូនដំណឹង៖ កប្បាសនៅតំបន់ខ្ពង់រាប និងកប្បាសកោះសមុទ្រ គឺជាដំណាំ tetraploid ដូច្នេះលទ្ធផលនៃការផ្ទុះជាញឹកញាប់នឹងជាការប្រកួតពីរ ឬច្រើន។កាលពីមុន ការប្រើ NAU cds ជាមូលដ្ឋានទិន្នន័យដើម្បីធ្វើការបំផ្ទុះទំនងជារកឃើញហ្សែនដូចគ្នាពីរដែលមានភាពខុសគ្នា SNP តិចតួចប៉ុណ្ណោះ។ជាធម្មតា ហ្សែនដូចគ្នាទាំងពីរមិនអាចបំបែកបានដោយការរចនាបឋមទេ ដូច្នេះពួកវាត្រូវបានចាត់ទុកដូចគ្នា។ប្រសិនបើមាន indel ជាក់ស្តែង primer ជាធម្មតាត្រូវបានរចនាឡើងនៅលើ indel ប៉ុន្តែនេះអាចនាំឱ្យរចនាសម្ព័ន្ធបន្ទាប់បន្សំនៃ primer ថាមពលទំនេរកាន់តែខ្ពស់ដែលនាំឱ្យមានការថយចុះនៃប្រសិទ្ធភាព amplification ប៉ុន្តែនេះគឺជៀសមិនរួច។
ការរកឃើញរចនាសម្ព័ន្ធបឋមបន្ទាប់បន្សំ៖
ជំហាន៖បើក oligo 7 → input template sequence → close sub-window → save → locate primer on template ចុច ctrl+D ដើម្បីកំណត់ប្រវែង primer → វិភាគរចនាសម្ព័ន្ធបន្ទាប់បន្សំផ្សេងៗ ដូចជា self-dimerization body, heterodimer, hairpin, mismatch, etc. រូបភាពពីរចុងក្រោយក្នុងរូបភាពទី 4 គឺជាលទ្ធផលតេស្តនៃ primers ។លទ្ធផលនៃ primer ខាងមុខគឺល្អ, មិនមានរចនាសម្ព័ន្ធ dimer ជាក់ស្តែងនិង hairpin, មិនមានមូលដ្ឋានបំពេញបន្ថែម, និងតម្លៃដាច់ខាតនៃថាមពលដោយឥតគិតថ្លៃគឺតិចជាង 4.5, ខណៈពេលដែល primer ខាងក្រោយបង្ហាញបន្តមូលដ្ឋាន 6 គឺបំពេញបន្ថែម, និងថាមពលដោយឥតគិតថ្លៃគឺ 8.8;លើសពីនេះទៀត dimer ធ្ងន់ធ្ងរជាងនេះលេចឡើងនៅចុងបញ្ចប់ 3′ ហើយ dimer នៃមូលដ្ឋាន 4 ជាប់គ្នាលេចឡើង។ទោះបីជាថាមពលឥតគិតថ្លៃមិនខ្ពស់ក៏ដោយ 3′ dimer Chl អាចប៉ះពាល់យ៉ាងធ្ងន់ធ្ងរដល់ភាពជាក់លាក់នៃការពង្រីក និងប្រសិទ្ធភាពនៃការពង្រីក។បន្ថែមពីលើនេះទៀត ចាំបាច់ត្រូវពិនិត្យមើលក្រវិលសក់ អេទីរ៉ូឌីមឺរ និងមិនស៊ីគ្នា។
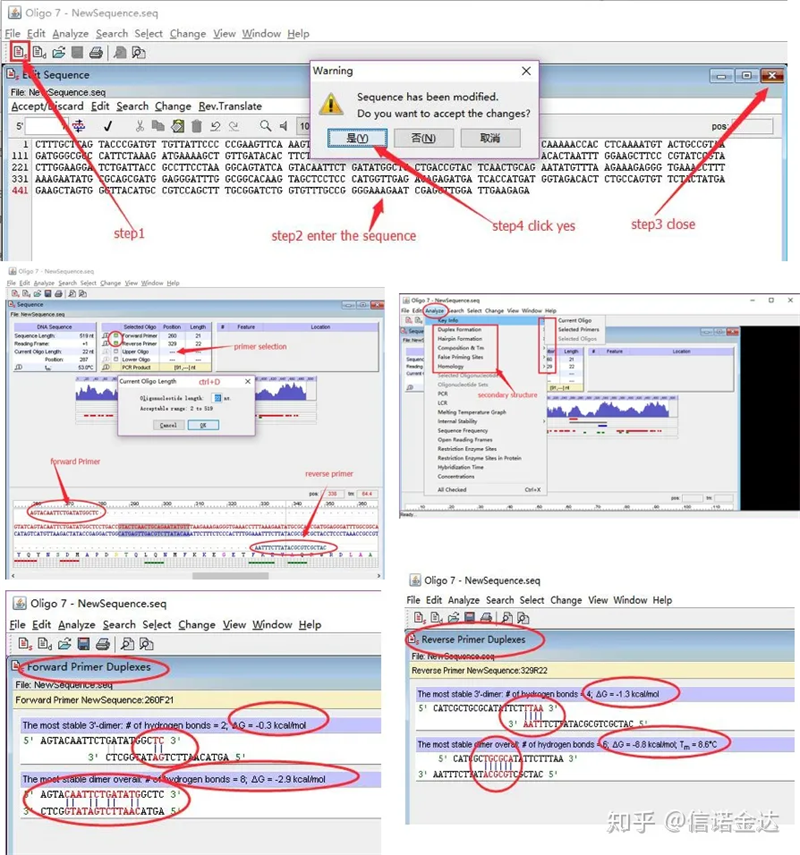
លទ្ធផលរកឃើញ Fig3 oligo7
ការរកឃើញប្រសិទ្ធភាពពង្រីក៖
ប្រសិទ្ធភាពពង្រីកនៃប្រតិកម្ម PCR ប៉ះពាល់យ៉ាងធ្ងន់ធ្ងរដល់លទ្ធផល PCR ។ផងដែរនៅក្នុង qRT-PCR ប្រសិទ្ធភាព amplification គឺមានសារៈសំខាន់ជាពិសេសសម្រាប់លទ្ធផលបរិមាណ។យកសារធាតុ ម៉ាស៊ីន និងពិធីការផ្សេងទៀតនៅក្នុងសតិបណ្ដោះអាសន្នប្រតិកម្ម។គុណភាពនៃ primers ក៏មានឥទ្ធិពលយ៉ាងខ្លាំងទៅលើប្រសិទ្ធភាព amplification នៃ qRT-PCR ផងដែរ។ដើម្បីធានាបាននូវភាពត្រឹមត្រូវនៃលទ្ធផល ទាំងបរិមាណ fluorescence ដែលទាក់ទង និងបរិមាណ fluorescence ដាច់ខាតចាំបាច់ត្រូវរកឃើញប្រសិទ្ធភាព amplification នៃ primers ។វាត្រូវបានគេទទួលស្គាល់ថាប្រសិទ្ធភាពពង្រីក qRT-PCR ដ៏មានប្រសិទ្ធភាពគឺចន្លោះពី 85% ទៅ 115% ។មានវិធីពីរយ៉ាង៖
1. វិធីសាស្ត្រខ្សែកោងស្តង់ដារ៖
ក.លាយ cDNA
ខ.ការរំលាយពណ៌ជម្រាល
c.qPCR
ឃ.សមីការតំរែតំរង់លីនេអ៊ែរ ដើម្បីគណនាប្រសិទ្ធភាពនៃការពង្រីក
2. LinRegPCR
LinRegPCR គឺជាកម្មវិធីសម្រាប់ការវិភាគទិន្នន័យ RT-PCR ពេលវេលាពិត ដែលគេហៅផងដែរថាទិន្នន័យ PCR បរិមាណ (qPCR) ដោយផ្អែកលើ SYBR Green ឬគីមីវិទ្យាស្រដៀងគ្នា។កម្មវិធីនេះប្រើទិន្នន័យកែតម្រូវដែលមិនមែនជាបន្ទាត់មូលដ្ឋាន អនុវត្តការកែតម្រូវបន្ទាត់មូលដ្ឋានលើគំរូនីមួយៗដោយឡែកពីគ្នា កំណត់វិនដូនៃលីនេអ៊ែរ ហើយបន្ទាប់មកប្រើការវិភាគតំរែតំរង់លីនេអ៊ែរដើម្បីឱ្យសមនឹងបន្ទាត់ត្រង់តាមរយៈសំណុំទិន្នន័យ PCR ។ពីជម្រាលនៃបន្ទាត់នេះប្រសិទ្ធភាព PCR នៃគំរូនីមួយៗត្រូវបានគណនា។ប្រសិទ្ធភាព PCR ជាមធ្យមក្នុងមួយអំពែក និងតម្លៃ Ct ក្នុងមួយសំណាកត្រូវបានប្រើដើម្បីគណនាកំហាប់ចាប់ផ្តើមក្នុងមួយសំណាក ដែលបង្ហាញក្នុងឯកតាហ្វ្លុយអូរីតាមអំពើចិត្ត។ការបញ្ចូលទិន្នន័យ និងលទ្ធផលគឺតាមរយៈសៀវភៅបញ្ជី Excel ។គំរូតែប៉ុណ្ណោះ
ការលាយគឺត្រូវបានទាមទារ, មិនមានជម្រាល
ជំហានត្រូវបានទាមទារ៖(យក Bole CFX96 ជាឧទាហរណ៍ មិនមែនម៉ាស៊ីនដែលមាន ABI ច្បាស់លាស់ទេ)
ពិសោធន៍៖វាគឺជាការពិសោធន៍ qPCR ស្តង់ដារ។
ទិន្នផលទិន្នន័យ qPCR៖LinRegPCR អាចស្គាល់ទម្រង់ពីរនៃឯកសារលទ្ធផល៖ RDML ឬ quantification លទ្ធផល Amplification ។តាមការពិត វាគឺជាតម្លៃនៃការរកឃើញក្នុងពេលជាក់ស្តែងនៃលេខវដ្ត និងសញ្ញា fluorescence ដោយម៉ាស៊ីន ហើយការពង្រីកត្រូវបានទទួលដោយការវិភាគតម្លៃនៃការផ្លាស់ប្តូរ fluorescence នៃប្រសិទ្ធភាពផ្នែកលីនេអ៊ែរ។
ការជ្រើសរើសទិន្នន័យ៖ តាមទ្រឹស្តី តម្លៃ RDML គួរតែអាចប្រើបាន។វាត្រូវបានប៉ាន់ស្មានថាបញ្ហានៃកុំព្យូទ័ររបស់ខ្ញុំគឺថាកម្មវិធីមិនអាចស្គាល់ RDML ដូច្នេះខ្ញុំមានតម្លៃលទ្ធផល Excel ជាទិន្នន័យដើម។វាត្រូវបានផ្ដល់អនុសាសន៍ឱ្យធ្វើការពិនិត្យទិន្នន័យយ៉ាងម៉ត់ចត់ជាមុនសិន ដូចជាការបរាជ័យនៃការបន្ថែមគំរូជាដើម។ ចំណុចអាចត្រូវបានលុបនៅក្នុងទិន្នន័យលទ្ធផល (ជាការពិតណាស់ អ្នកមិនអាចលុបពួកវាបានទេ LinRegPCR នឹងមិនអើពើចំណុចទាំងនេះនៅដំណាក់កាលក្រោយ)
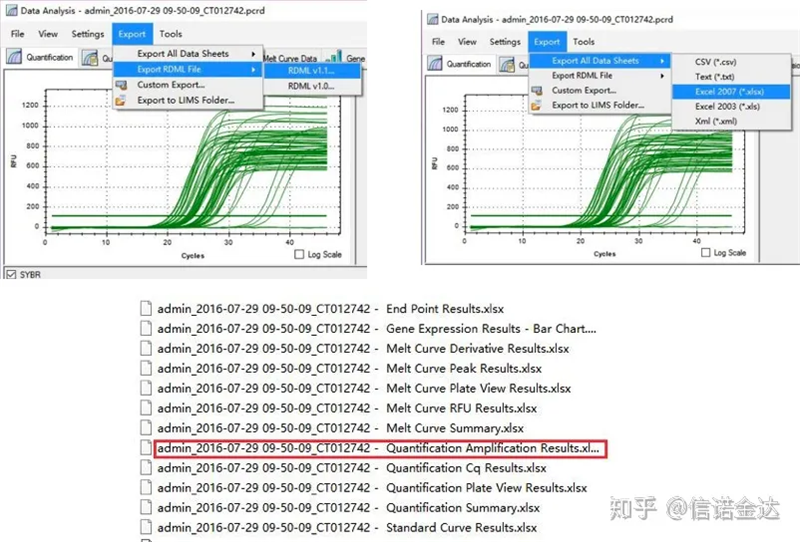
ការនាំចេញទិន្នន័យ fig5 qPCR
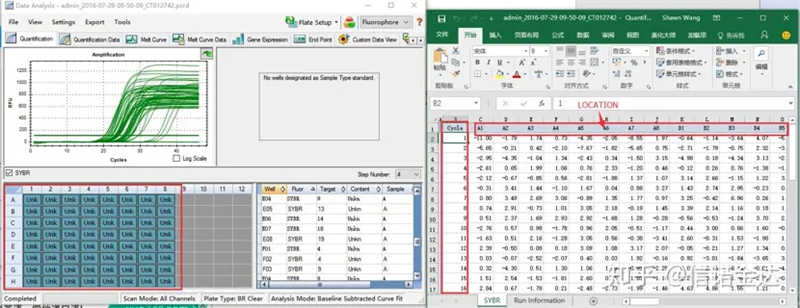
រូបភាពទី 6 ការជ្រើសរើសគំរូបេក្ខជន
ការបញ្ចូលទិន្នន័យ៖បើក qualification amplification results.xls, → បើក LinRegPCR → ឯកសារ → អានពី excel → ជ្រើសរើសប៉ារ៉ាម៉ែត្រដូចបង្ហាញក្នុងរូបភាពទី 7 → យល់ព្រម → ចុចកំណត់បន្ទាត់មូលដ្ឋាន
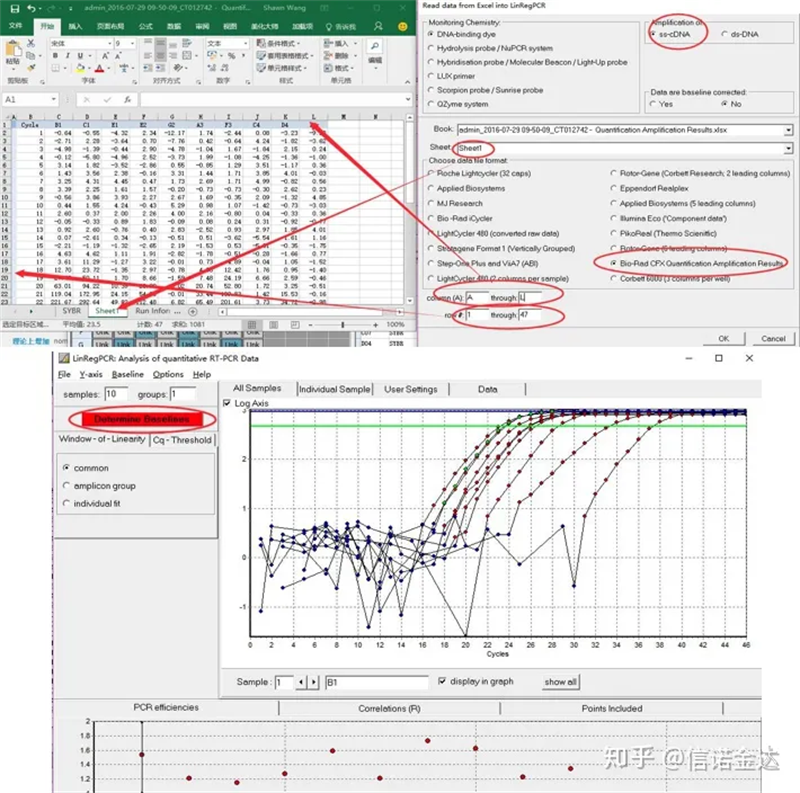
Fig7 ជំហាននៃការបញ្ចូលទិន្នន័យ linRegPCR
លទ្ធផល៖ប្រសិនបើមិនមានពាក្យដដែលៗទេ មិនចាំបាច់ដាក់ក្រុមទេ។ប្រសិនបើមានពាក្យដដែលៗ ការដាក់ជាក្រុមអាចត្រូវបានកែសម្រួលក្នុងការដាក់ជាក្រុមគំរូ ហើយឈ្មោះហ្សែនត្រូវបានបញ្ចូលក្នុងឧបករណ៍កំណត់អត្តសញ្ញាណ ហើយបន្ទាប់មកហ្សែនដូចគ្នានឹងត្រូវដាក់ជាក្រុមដោយស្វ័យប្រវត្តិ។ចុងក្រោយចុចលើឯកសារ នាំចេញ excel និងមើលលទ្ធផល។ប្រសិទ្ធភាពពង្រីក និងលទ្ធផល R2 នៃអណ្តូងនីមួយៗនឹងត្រូវបានបង្ហាញ។ទីពីរ ប្រសិនបើអ្នកបែងចែកជាក្រុម ប្រសិទ្ធភាពនៃការពង្រីកមធ្យមដែលបានកែតម្រូវនឹងត្រូវបានបង្ហាញ។ត្រូវប្រាកដថាប្រសិទ្ធភាពពង្រីកនៃ primer នីមួយៗមានចន្លោះពី 85% ទៅ 115%។ប្រសិនបើវាធំពេក ឬតូចពេក វាមានន័យថា ប្រសិទ្ធភាពពង្រីកនៃ primer គឺអន់។
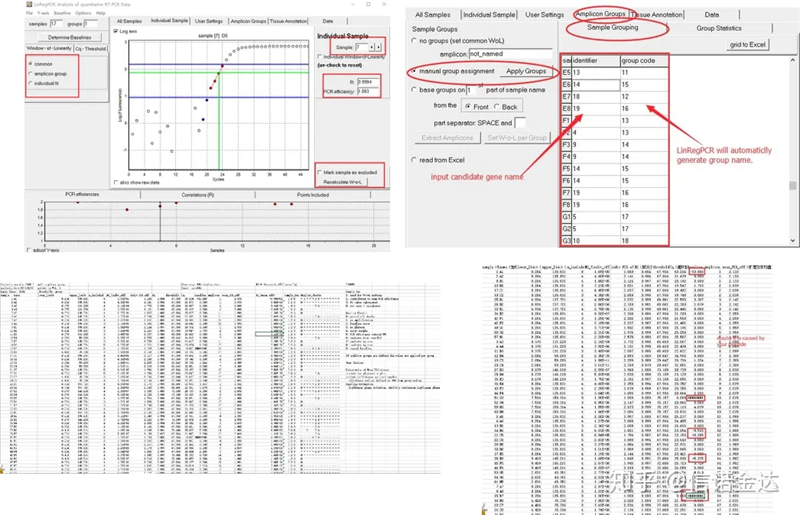
រូបភាពទី ៨ លទ្ធផល និងទិន្នន័យ
ដំណើរការពិសោធន៍៖
តម្រូវការគុណភាព RNA៖
ភាពបរិសុទ្ធ:១.៧2.0 បង្ហាញថាអាចមាន isothiocyanate ដែលនៅសល់។អាស៊ីត nucleic ស្អាត A260/A230 គួរតែមានប្រហែល 2 ។ប្រសិនបើមានការស្រូបយកខ្លាំងនៅ 230 nm វាបង្ហាញថាមានសមាសធាតុសរីរាង្គដូចជា phenate ions ។លើសពីនេះទៀតវាអាចត្រូវបានរកឃើញដោយ 1.5% agarose gel electrophoresis ។ចង្អុលសញ្ញាសម្គាល់ ពីព្រោះ ssRNA មិនមាន denaturation ហើយលោការីតទម្ងន់ម៉ូលេគុលមិនមានទំនាក់ទំនងលីនេអ៊ែរទេ ហើយទម្ងន់ម៉ូលេគុលមិនអាចត្រូវបានបង្ហាញបានត្រឹមត្រូវ។ការផ្តោតអារម្មណ៍៖ តាមទ្រឹស្តីទេ។តិចជាង 100ng/ul ប្រសិនបើកំហាប់ទាបពេក ភាពបរិសុទ្ធជាទូទៅទាប មិនខ្ពស់ទេ។
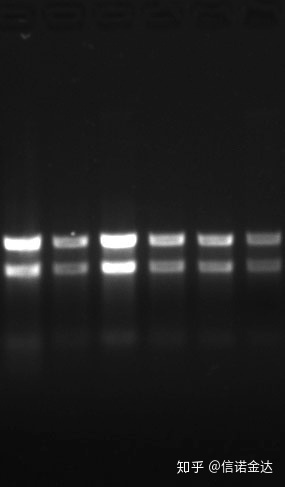
រូបភាពទី 9 RNA ជែល
លើសពីនេះទៀត ប្រសិនបើសំណាកមានតម្លៃ ហើយកំហាប់ RNA ខ្ពស់ វាត្រូវបានណែនាំអោយ aliquot វាបន្ទាប់ពីការស្រង់ចេញ ហើយ dilute RNA ទៅកំហាប់ចុងក្រោយនៃ 100-300ng/ul សម្រាប់ការចម្លងបញ្ច្រាស។ក្នុងដំណើរការនៃការចម្លងបញ្ច្រាសនៅពេលដែល mRNA ត្រូវបានចម្លង សារធាតុ primers oligo (dt) ដែលអាចភ្ជាប់ជាពិសេសទៅនឹងកន្ទុយ polyA ត្រូវបានប្រើសម្រាប់ការចម្លងបញ្ច្រាស ចំណែកឯ lncRNA និង circRNA ប្រើ primers ចៃដន្យ hexamer (Random 6 mer) សម្រាប់ការចម្លងបញ្ច្រាសនៃ RNA សរុបសម្រាប់ miRNA, miRNA-specific miRNA-specific neck-looprs ត្រូវបានប្រើសម្រាប់ការចម្លងឡើងវិញនៃ neck-loopersឥឡូវនេះក្រុមហ៊ុនជាច្រើនបានដាក់ចេញនូវឧបករណ៍កាត់កន្ទុយពិសេស។សម្រាប់វិធី Stem-loop វិធីសាស្ត្រកាត់កន្ទុយគឺងាយស្រួលជាង ទិន្នផលខ្ពស់ និងការសន្សំសំចៃ reagent ប៉ុន្តែឥទ្ធិពលនៃការបែងចែក miRNAs នៃគ្រួសារតែមួយមិនគួរល្អដូចវិធី Stem-loop ទេ។ឧបករណ៍ចម្លងបញ្ច្រាសនីមួយៗមានតម្រូវការសម្រាប់ការប្រមូលផ្តុំនៃ primers ជាក់លាក់នៃហ្សែន (stem-loops) ។ឯកសារយោងខាងក្នុងដែលប្រើសម្រាប់ miRNA គឺ U6 ។នៅក្នុងដំណើរការនៃការដាក់បញ្ច្រាស់ដើម បំពង់ U6 គួរតែត្រូវបានដាក់បញ្ច្រាសដោយឡែកពីគ្នា ហើយទ្រនាប់ខាងមុខ និងខាងក្រោយរបស់ U6 គួរតែត្រូវបានបន្ថែមដោយផ្ទាល់។ទាំង circRNA និង lncRNA អាចប្រើ HKGs ជាឯកសារយោងខាងក្នុង។ក្នុងការរកឃើញ cDNA,
ប្រសិនបើមិនមានបញ្ហាជាមួយ RNA នោះ cDNA ក៏គួរតែល្អដែរ។ទោះជាយ៉ាងណាក៏ដោយ ប្រសិនបើភាពល្អឥតខ្ចោះនៃការពិសោធន៍ត្រូវបានបន្ត វាជាការល្អបំផុតក្នុងការប្រើហ្សែនយោងខាងក្នុង (ហ្សែនយោង, RG) ដែលអាចបែងចែក gDNA ពីស៊ីឌី។ជាទូទៅ RG គឺជាហ្សែនថែរក្សាផ្ទះ។, HKG) ដូចបង្ហាញក្នុងរូបភាពទី 10;នៅពេលនោះ ខ្ញុំកំពុងបង្កើតប្រូតេអ៊ីនស្តុកសណ្តែកសៀង ហើយបានប្រើ actin7 ដែលមានផ្ទុក introns ជាឯកសារយោងខាងក្នុង។ទំហំនៃបំណែកដែលបានពង្រីកនៃ primer នេះនៅក្នុង gDNA គឺ 452bp ហើយប្រសិនបើ cDNA ត្រូវបានប្រើជាគំរូ វាគឺ 142bp ។បន្ទាប់មកលទ្ធផលតេស្តបានរកឃើញថាផ្នែកមួយនៃ cDNA ពិតជាត្រូវបានបំពុលដោយ gDNA ហើយវាក៏បង្ហាញផងដែរថាមិនមានបញ្ហាជាមួយនឹងលទ្ធផលនៃការចម្លងបញ្ច្រាសទេ ហើយវាអាចត្រូវបានប្រើជាគំរូសម្រាប់ PCR ។វាគ្មានប្រយោជន៍ទេក្នុងការដំណើរការ agarose gel electrophoresis ដោយផ្ទាល់ជាមួយ cDNA ហើយវាគឺជាក្រុមដែលសាយភាយ ដែលមិនគួរឱ្យជឿ។
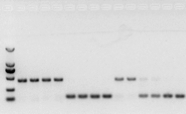
រូបភាពទី 10 ការរកឃើញ cDNA
ការកំណត់លក្ខខណ្ឌ qPCRជាទូទៅមិនមានបញ្ហាអ្វីទេយោងទៅតាមពិធីការនៃឧបករណ៍នេះ ជាចម្បងនៅក្នុងជំហាននៃតម្លៃ tm ។ប្រសិនបើ primers មួយចំនួនមិនត្រូវបានរចនាយ៉ាងល្អកំឡុងពេលរចនា primer ដែលបណ្តាលឱ្យមានភាពខុសគ្នាខ្លាំងរវាងតម្លៃ tm និងទ្រឹស្តី 60°C វាត្រូវបានណែនាំថា cDNA បន្ទាប់ពីសំណាកត្រូវបានលាយបញ្ចូលគ្នា ដំណើរការ PCR gradient ជាមួយ primers ហើយព្យាយាមជៀសវាងការកំណត់សីតុណ្ហភាពដោយគ្មាន bands ជាតម្លៃ TM ។
ការវិភាគទិន្នន័យ
វិធីសាស្រ្តដំណើរការ PCR បរិមាណ fluorescence ទាក់ទងធម្មតាគឺផ្អែកលើ 2-ΔΔCT.គំរូដំណើរការទិន្នន័យ។
ផលិតផលដែលពាក់ព័ន្ធ៖
ពេលវេលាពិត PCR ងាយស្រួលTM - តាកម៉ាន់
ពេលវេលាពិត PCR ងាយស្រួលTM -SYBR GREEN I
RT Easy I (Master Premix សម្រាប់ការសំយោគ cDNA strand ដំបូង)
RT Easy II (Master Premix សម្រាប់ការសំយោគ cDNA strand ដំបូងសម្រាប់ qPCR)
ពេលវេលាផ្សាយ៖ ថ្ងៃទី ១៤ ខែមីនា ឆ្នាំ ២០២៣



