



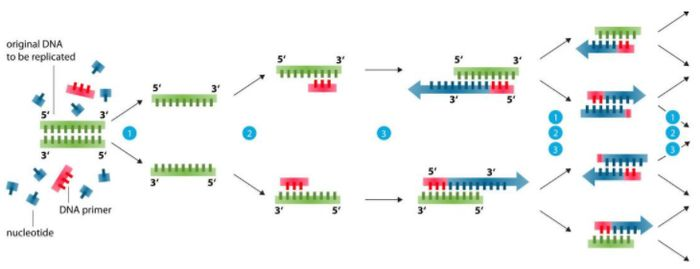
មូលដ្ឋានគ្រឹះនៃការរចនាបឋម (99% បញ្ហាអាចត្រូវបានដោះស្រាយ)
1. ប្រវែងបឋម៖ សៀវភៅសិក្សាត្រូវការ 15-30bp ជាធម្មតាប្រហែល 20bp ។លក្ខខណ្ឌជាក់ស្តែងគឺល្អប្រសើរជាងមុនដើម្បីឱ្យមាន 18-24bp ដើម្បីធានាបាននូវភាពជាក់លាក់ប៉ុន្តែយូរជាងនេះកាន់តែប្រសើរ primer វែងពេកក៏នឹងកាត់បន្ថយភាពជាក់លាក់និងកាត់បន្ថយទិន្នផលផងដែរ។
2. Primer amplification span: 200-500bp គឺសមរម្យ ហើយបំណែកអាចពង្រីកដល់ 10kb ក្រោមលក្ខខណ្ឌជាក់លាក់។
3. មូលដ្ឋានបឋម៖ ខ្លឹមសារនៃ G+C គួរតែមាន 40-60%, ឥទ្ធិពលពង្រីក G+C តិចតួចពេកគឺមិនល្អទេ G+C ច្រើនពេកងាយនឹងបង្ហាញក្រុមមិនជាក់លាក់។ATGC ត្រូវបានចែកចាយយ៉ាងល្អបំផុតដោយចៃដន្យ ដោយជៀសវាងចង្កោមនៃ purine ឬ pyrimidine nucleotides ច្រើនជាង 5 ។Multi-gc សម្រាប់លំដាប់ 5′ និងកម្រិតមធ្យមដើម្បីបង្កើនស្ថេរភាព ជៀសវាង GC សម្បូរបែបនៅចុងបញ្ចប់ 3′ គ្មាន GC សម្រាប់មូលដ្ឋាន 3 ចុងក្រោយ ឬគ្មាន GC សម្រាប់ 3 នៃមូលដ្ឋាន 5 ចុងក្រោយ។
4. ជៀសវាងរចនាសម្ព័ន្ធបន្ទាប់បន្សំនៅក្នុង primers ហើយជៀសវាងការបំពេញបន្ថែមរវាង primers ពីរ ជាពិសេសការបំពេញបន្ថែមនៅចុង 3' បើមិនដូច្នេះទេ primer dimer នឹងត្រូវបានបង្កើតឡើង ហើយក្រុម amplified មិនជាក់លាក់នឹងត្រូវបានបង្កើត។
5. មូលដ្ឋាននៅចុង 3' នៃ primers ជាពិសេសមូលដ្ឋានចុងក្រោយ និង penultimate គួរតែត្រូវបានផ្គូផ្គងយ៉ាងតឹងរ៉ឹង ដើម្បីជៀសវាងការបរាជ័យ PCR ដោយសារតែមូលដ្ឋានស្ថានីយដែលមិនបានផ្គូផ្គង។
6. Primers មាន ឬអាចត្រូវបានបន្ថែមជាមួយនឹងកន្លែងបំបែកដែលសមរម្យ ហើយលំដាប់គោលដៅពង្រីកគួរតែមានកន្លែងបំបែកត្រឹមត្រូវ ដែលមានប្រយោជន៍ខ្លាំងណាស់សម្រាប់ការវិភាគការបំបែកឬការក្លូនម៉ូលេគុល។
7. ភាពជាក់លាក់នៃ primers: primers មិនគួរមានភាពដូចគ្នាជាក់ស្តែងជាមួយនឹងលំដាប់ផ្សេងទៀតនៅក្នុង database លំដាប់អាស៊ីត nucleic។
8. រៀនប្រើកម្មវិធី៖ PP5, Oligo6, DNAstar, Vector NTI, primer3 (ការរចនាតាមអ៊ីនធឺណិតនេះដំណើរការល្អបំផុត)។
ខ្លឹមសារខាងលើអាចដោះស្រាយបានយ៉ាងហោចណាស់ 99% នៃបញ្ហាការរចនាបឋម។
គ្រប់គ្រងព័ត៌មានលម្អិតនៃការរចនាបឋម
1. ប្រវែង primer
ប្រវែង primer ទូទៅគឺ 18-30 មូលដ្ឋាន។ជាទូទៅកត្តាសំខាន់បំផុតដែលកំណត់សីតុណ្ហភាពនៃការស្រោបនៃ primer គឺប្រវែងនៃ primer ។សីតុណ្ហភាព annealing នៃ primer ត្រូវបានជ្រើសរើសជាទូទៅ (តម្លៃ Tm -5 ℃) ហើយខ្លះប្រើតម្លៃ Tm ដោយផ្ទាល់។រូបមន្តខាងក្រោមអាចត្រូវបានប្រើដើម្បីគណនាប្រហែលសីតុណ្ហភាព annealing នៃ primers ។
នៅពេលដែលប្រវែង primer តិចជាង 20bp: [4(G+C)+2(A+T)]-5℃
នៅពេលដែលប្រវែង primer ធំជាង 20bp: 62.3℃+0.41℃(%GC)-500/length-5℃
លើសពីនេះទៀតកម្មវិធីជាច្រើនក៏អាចត្រូវបានប្រើដើម្បីគណនាសីតុណ្ហភាព annealing គោលការណ៍គណនានឹងខុសគ្នាដូច្នេះជួនកាលតម្លៃដែលបានគណនាអាចមានគម្លាតតូចមួយ។ដើម្បីបង្កើនប្រសិទ្ធភាពប្រតិកម្ម PCR ថ្នាំ primers ខ្លីបំផុតដែលធានានូវសីតុណ្ហភាពមិនតិចជាង 54 ℃ ត្រូវបានប្រើសម្រាប់ប្រសិទ្ធភាព និងជាក់លាក់ល្អបំផុត។
សរុបមក ភាពជាក់លាក់នៃ primer កើនឡើងដោយកត្តាបួនសម្រាប់ nucleotide បន្ថែមនីមួយៗ ដូច្នេះប្រវែង primer អប្បបរមាសម្រាប់កម្មវិធីភាគច្រើនគឺ 18 nucleotides។ដែនកំណត់ខាងលើនៃប្រវែង primer គឺមិនសូវសំខាន់ទេ ភាគច្រើនទាក់ទងនឹងប្រសិទ្ធភាពប្រតិកម្ម។ដោយសារតែ entropy កាន់តែយូរ primer កាន់តែទាប អត្រាដែលវាភ្ជាប់ទៅ DNA គោលដៅ ដើម្បីបង្កើតជាគំរូខ្សែពីរដែលមានស្ថេរភាពសម្រាប់ DNA polymerase ដើម្បីចង។
នៅពេលប្រើកម្មវិធីដើម្បីរចនា primers ប្រវែងនៃ primers អាចត្រូវបានកំណត់ដោយតម្លៃ TM ជាវេន ជាពិសេសសម្រាប់ primers នៃ fluorescence quantitative PCR, TM=60 ℃ ឬដូច្នេះគួរតែត្រូវបានគ្រប់គ្រង។
2.GC មាតិកា
ជាទូទៅមាតិកានៃ G + C នៅក្នុងលំដាប់បឋមគឺ 40% ~ 60% ហើយមាតិកា GC និងតម្លៃ Tm នៃ primer មួយគូគួរតែត្រូវបានសម្របសម្រួល។ប្រសិនបើ primer មានទំនោរ GC ឬ AT ធ្ងន់ធ្ងរ បរិមាណសមស្របនៃកន្ទុយ A, T ឬ G និង C អាចត្រូវបានបន្ថែមទៅចុង 5' នៃ primer ។
3. សីតុណ្ហភាព Annealing
សីតុណ្ហភាព annealing គួរតែមាន 5 ℃ទាបជាងសីតុណ្ហភាព unchain ។ប្រសិនបើចំនួននៃមូលដ្ឋាន primer មានតិចតួចនោះសីតុណ្ហភាព annealing អាចត្រូវបានកើនឡើងយ៉ាងសមរម្យដែលអាចបង្កើនភាពជាក់លាក់នៃ PCR ។ប្រសិនបើចំនួននៃមូលដ្ឋានមានទំហំធំនោះសីតុណ្ហភាព annealing អាចត្រូវបានកាត់បន្ថយសមរម្យ។ភាពខុសគ្នានៃសីតុណ្ហភាព annealing រវាង primers មួយគូនៃ 4 ℃ ~ 6 ℃ នឹងមិនប៉ះពាល់ដល់ទិន្នផល PCR នោះទេប៉ុន្តែតាមឧត្ដមគតិសីតុណ្ហភាព annealing នៃ primers មួយគូគឺដូចគ្នាដែលអាចប្រែប្រួលរវាង 55 ℃ ~ 75 ℃។
4. ជៀសវាងតំបន់រចនាសម្ព័ន្ធបន្ទាប់បន្សំនៃគំរូ amplification
វាជាការល្អបំផុតដើម្បីជៀសវាងតំបន់រចនាសម្ព័ន្ធបន្ទាប់បន្សំនៃគំរូនៅពេលជ្រើសរើសបំណែកពង្រីក។រចនាសម្ព័ន្ធបន្ទាប់បន្សំដែលមានស្ថេរភាពនៃបំណែកគោលដៅអាចត្រូវបានព្យាករណ៍ និងប៉ាន់ស្មានដោយកម្មវិធីកុំព្យូទ័រដែលពាក់ព័ន្ធ ដែលមានប្រយោជន៍សម្រាប់ការជ្រើសរើសគំរូ។លទ្ធផលពិសោធន៍បង្ហាញថាការពង្រីកនេះច្រើនតែមិនជោគជ័យនៅពេលដែលថាមពលទំនេរ (△G) នៃតំបន់ដែលត្រូវពង្រីកគឺតិចជាង 58.6lkJ/mol។
5. មិនត្រូវគ្នាជាមួយ DNA គោលដៅ
នៅពេលដែលលំដាប់ DNA គោលដៅពង្រីកមានទំហំធំ សារធាតុ primer អាចភ្ជាប់ទៅនឹងផ្នែកជាច្រើននៃ DNA គោលដៅ ដែលបណ្តាលឱ្យមានក្រុមជាច្រើនលេចឡើងនៅក្នុងលទ្ធផល។លើកនេះចាំបាច់ត្រូវប្រើការសាកល្បងកម្មវិធី BLAST គេហទំព័រ៖http://www.ncbi.nlm.nih.gov/BLAST/.ជ្រើសរើសតម្រឹមលំដាប់ពីរ (bl2seq) ។
ការបិទភ្ជាប់លំដាប់បឋមទៅតំបន់ 1 និងតម្រង់ជួរ DNA ទៅតំបន់ 2 គឺអាចផ្លាស់ប្តូរបាន ហើយ BLAST គណនាការបំពេញបន្ថែម ការប្រឆាំង និងលទ្ធភាពផ្សេងទៀត ដូច្នេះអ្នកប្រើប្រាស់មិនចាំបាច់កត់សំគាល់ថាតើខ្សែសង្វាក់ទាំងពីរជាខ្សែសង្វាក់អារម្មណ៍នោះទេ។អ្នកក៏អាចបញ្ចូលលេខ GI ប្រសិនបើអ្នកស្គាល់លេខ GI នៃលំដាប់នៅក្នុងមូលដ្ឋានទិន្នន័យ ដូច្នេះអ្នកមិនចាំបាច់បិទភ្ជាប់ផ្នែកធំនៃលំដាប់នោះទេ។ជាចុងក្រោយ សូមចុចតម្រឹមនៅលេខ 3 ដើម្បីមើលថាតើ primer មានកន្លែងដូចគ្នាច្រើននៅក្នុង DNA គោលដៅដែរឬទេ។
6. ស្ថានីយ primer
ចុង 3 'នៃ primer គឺជាកន្លែងដែលផ្នែកបន្ថែមចាប់ផ្តើម ដូច្នេះវាជាការសំខាន់ក្នុងការទប់ស្កាត់ភាពមិនស៊ីគ្នាពីការចាប់ផ្តើមនៅទីនោះ។ចុង 3 'មិនគួរលើសពី 3 ជាប់គ្នា G ឬ C ទេ ពីព្រោះវានឹងធ្វើឱ្យ primer ត្រូវបានបង្កឡើងដោយច្រឡំនៅក្នុងតំបន់លំដាប់នៃ G+C ។ចុង 3' មិនអាចបង្កើតរចនាសម្ព័ន្ធបន្ទាប់បន្សំណាមួយបានទេ លើកលែងតែក្នុងប្រតិកម្ម PCR ពិសេស (AS-PCR) ចុងបញ្ចប់ 3′ នៃ primer មិនអាចត្រូវគ្នាបានទេ។ឧទាហរណ៍ ប្រសិនបើតំបន់បំប្លែងកូដត្រូវបានពង្រីកនោះ ចុង 3 'នៃ primer មិនគួរត្រូវបានបញ្ចប់នៅទីតាំងទីបីនៃ codon ទេ ព្រោះទីតាំងទីបីនៃ codon ងាយនឹងខូច ដែលនឹងប៉ះពាល់ដល់ភាពជាក់លាក់ និងប្រសិទ្ធភាពនៃការពង្រីក។នៅពេលប្រើថ្នាំ primers ឧបសម្ព័ន្ធ សូមមើលតារាងប្រើ codon យកចិត្តទុកដាក់លើចំណូលចិត្តជីវសាស្រ្ត កុំប្រើ primers ភ្ជាប់នៅចុង 3′ ហើយប្រើកំហាប់ខ្ពស់នៃ primers (1uM-3uM)។
7. រចនាសម្ព័ន្ធបន្ទាប់បន្សំនៃ primers
primers ខ្លួនឯងមិនគួរមានបន្តបន្ទាប់គ្នាទេ បើមិនដូច្នេះទេ primers ខ្លួនឯងនឹងបត់ចូលទៅក្នុងរចនាសម្ព័ន្ធ hairpin ហើយរចនាសម្ព័ន្ធបន្ទាប់បន្សំនេះនឹងប៉ះពាល់ដល់ការចងនៃ primers និង templates ដោយសារតែការរាំងស្ទះ steric ។ប្រសិនបើការវិនិច្ឆ័យសិប្បនិម្មិតត្រូវបានប្រើ មូលដ្ឋានបន្ថែមជាបន្តបន្ទាប់នៃ primers ខ្លួនឯងមិនគួរធំជាង 3bp ទេ។មិនគួរមានការបំពេញបន្ថែមរវាង primer ទាំងពីរទេ ជាពិសេសការត្រួតគ្នានៃចុង 3 ' គួរតែត្រូវបានជៀសវាងដើម្បីការពារការបង្កើត primer dimers ។ជាទូទៅ វាមិនគួរមានច្រើនជាង 4 មូលដ្ឋានជាប់គ្នា ឬភាពដូចគ្នារវាង primers មួយគូ។
8. បន្ថែមសញ្ញាសម្គាល់ ឬទីតាំង
ចុង 5 'មានឥទ្ធិពលតិចតួចលើភាពជាក់លាក់នៃ amplification ហើយដូច្នេះអាចត្រូវបានកែប្រែដោយមិនប៉ះពាល់ដល់ភាពជាក់លាក់នៃ amplification ។ការកែប្រែនៃ primer 5' end រួមបញ្ចូល: ការបន្ថែមកន្លែងដាក់កម្រិតអង់ស៊ីម;ដាក់ស្លាក biotin, fluorescence, digoxin, Eu3+ ជាដើម។ ណែនាំពីលំដាប់ DNA ដែលភ្ជាប់ប្រូតេអ៊ីន;ការណែនាំកន្លែងផ្លាស់ប្តូរ ការបញ្ចូល និងបាត់លំដាប់បំរែបំរួល និងការណែនាំអំពីលំដាប់ផ្សព្វផ្សាយ។លំដាប់បន្ថែមដែលមិនមាននៅលើលំដាប់គោលដៅ ដូចជាគេហទំព័រដាក់កម្រិត និងលំដាប់ផ្សព្វផ្សាយ អាចត្រូវបានបន្ថែមទៅចុង 5′ នៃ primer ដោយមិនប៉ះពាល់ដល់ភាពជាក់លាក់។លំដាប់ទាំងនេះមិនត្រូវបានរាប់បញ្ចូលក្នុងការគណនាតម្លៃ primer Tm ទេ ប៉ុន្តែគួរតែត្រូវបានសាកល្បងសម្រាប់ការបំពេញបន្ថែម និងរចនាសម្ព័ន្ធបន្ទាប់បន្សំខាងក្នុង។
9. ក្លូនរង
ភាគច្រើន PCR គ្រាន់តែជាការក្លូនបឋមប៉ុណ្ណោះ ហើយបន្ទាប់មកយើងត្រូវបញ្ចូលបំណែកគោលដៅទៅជាវ៉ិចទ័រផ្សេងៗ ដូច្នេះយើងត្រូវរចនាមូលដ្ឋានបន្ថែមសម្រាប់ប្រតិបត្តិការបន្ទាប់ក្នុងជំហាន PCR ។
លំដាប់មួយចំនួនដែលបានរចនាឡើងសម្រាប់ subcloning ត្រូវបានសង្ខេបដូចខាងក្រោម។
គេហទំព័ររឹតបន្តឹង endonuclease ត្រូវបានបន្ថែម
ការបន្ថែមកន្លែងដាក់កម្រិតអង់ស៊ីមគឺជាវិធីសាស្រ្តដែលប្រើជាទូទៅបំផុតសម្រាប់ subcloning ផលិតផល PCR ។ជាទូទៅ កន្លែងបំបែកគឺ 6 មូលដ្ឋាន បន្ថែមពីលើ 5 'ចុងបញ្ចប់នៃកន្លែងបំបែកត្រូវបន្ថែមមូលដ្ឋានការពារ 2 ~ 3 ។ទោះជាយ៉ាងណាក៏ដោយចំនួននៃមូលដ្ឋានការពារដែលត្រូវការដោយអង់ស៊ីមផ្សេងៗគ្នាគឺខុសគ្នា។ឧទាហរណ៍ SalⅠ មិនត្រូវការមូលដ្ឋានការពារ EcoRⅤ ទាមទារមូលដ្ឋានការពារ 1 NotⅠ ទាមទារមូលដ្ឋានការពារ 2 និង Hind Ⅲ ត្រូវការមូលដ្ឋានការពារ 3 ។
LIC បន្ថែមកន្ទុយ
ឈ្មោះពេញរបស់ LIC គឺ Ligation-Independent cloning ដែលជាវិធីសាស្ត្រក្លូនដែលត្រូវបានបង្កើតឡើងដោយ Navogen ជាពិសេសសម្រាប់ផ្នែករបស់វានៃវ៉ិចទ័រ pET ។ក្រុមហ៊ុនដឹកជញ្ជូន pET ដែលរៀបចំដោយវិធីសាស្ត្រ LIC មាន 12-15 base single strand sticky ដែលមិនបំពេញបន្ថែម ដែលបំពេញបន្ថែមចុងស្អិតដែលត្រូវគ្នានៅលើបំណែកបញ្ចូលគោលដៅ។សម្រាប់គោលបំណងពង្រីក លំដាប់បឋម 5′ នៃបំណែកដែលបានបញ្ចូលគួរតែបំពេញវ៉ិចទ័រ LIC ។សកម្មភាពបន្ថែមនៃ 3′→5′ នៃ T4 DNA polymerase អាចបង្កើតជាកំណាត់ស្អិតតែមួយនៅលើបំណែកដែលបានបញ្ចូលបន្ទាប់ពីរយៈពេលខ្លី។ដោយសារតែផលិតផលអាចត្រូវបានបង្កើតឡើងបានតែពីការ annealing ទៅវិញទៅមកនៃបំណែកបញ្ចូលដែលបានរៀបចំនិងវ៉ិចទ័រ, វិធីសាស្រ្តនេះគឺលឿននិងមានប្រសិទ្ធិភាពខ្លាំងណាស់ហើយវាត្រូវបានដឹកនាំការក្លូន។
ការណែនាំ TA ក្លូនបន្ថែមកន្ទុយ
ការក្លូន TA មិនអាចកំណត់គោលដៅបំណែកទៅជាវ៉ិចទ័របានទេ ដូច្នេះក្រោយមក Invitrogen បានណែនាំវ៉ិចទ័រដែលអាចកំណត់គោលដៅការក្លូន ដែលមានមូលដ្ឋាន GTGGS លេចធ្លោចំនួនបួននៅចុងម្ខាង។ដូច្នេះនៅក្នុងការរចនានៃ PCR primers លំដាប់បំពេញបន្ថែមគួរតែត្រូវបានបន្ថែមទៅតាមនោះ ដូច្នេះបំណែកអាចត្រូវបាន "តម្រង់ទិស" ។
ប្រសិនបើអ្នកខ្វះពេលវេលា អ្នកអាចសាកល្បងការសំយោគដោយផ្ទាល់ ដោយរួមបញ្ចូលគ្នានូវហ្សែនជាមួយនឹងវ៉ិចទ័រ ដែលជាអ្វីដែលយើងហៅថា ការសំយោគហ្សែន ET នៅក្នុង musecularists ។
ឃ. វិធីសាស្ត្រក្លូន In-Fusion
មិនតម្រូវឱ្យមាន ligase មិនត្រូវការប្រតិកម្មយូរទេ។ដរាបណាលំដាប់មួយនៅចុងទាំងពីរនៃវ៉ិចទ័រលីនេអ៊ែរត្រូវបានណែនាំនៅក្នុងការរចនានៃ primers បន្ទាប់មកផលិតផល PCR និងវ៉ិចទ័រលីនេអ៊ែរត្រូវបានបញ្ចូលទៅក្នុងដំណោះស្រាយអង់ស៊ីម in-fusion ដែលមាន BSA ហើយដាក់នៅសីតុណ្ហភាពបន្ទប់រយៈពេលកន្លះម៉ោង ការផ្លាស់ប្តូរអាចត្រូវបានអនុវត្ត។វិធីសាស្រ្តនេះគឺសមរម្យជាពិសេសសម្រាប់ការបំប្លែងទំហំធំ។
10. លាយ primer
ពេលខ្លះ មានតែព័ត៌មានលំដាប់លំដោយកំណត់ប៉ុណ្ណោះដែលត្រូវបានដឹងអំពីការរចនាបឋម។ឧទាហរណ៍ ប្រសិនបើគេស្គាល់តែលំដាប់អាស៊ីដអាមីណូប៉ុណ្ណោះនោះ សារធាតុ primer រួមបញ្ចូលគ្នាអាចត្រូវបានរចនាឡើង។ថ្នាំ primer រួមបញ្ចូលគ្នាគឺជាល្បាយនៃលំដាប់ផ្សេងគ្នាដែលតំណាងឱ្យលទ្ធភាពមូលដ្ឋានផ្សេងគ្នាទាំងអស់ដែលអ៊ិនកូដអាស៊ីតអាមីណូតែមួយ។ដើម្បីបង្កើនភាពជាក់លាក់ អ្នកអាចយោងទៅលើតារាងការប្រើប្រាស់ codon ដើម្បីកាត់បន្ថយការភ្ជាប់គ្នាដោយយោងទៅតាមចំណូលចិត្តការប្រើប្រាស់មូលដ្ឋាននៃសារពាង្គកាយផ្សេងៗគ្នា។Hypoxanthine អាចត្រូវបានផ្គូផ្គងជាមួយនឹងមូលដ្ឋានទាំងអស់ដើម្បីកាត់បន្ថយសីតុណ្ហភាព annealing នៃ primer ។កុំប្រើមូលដ្ឋានដែលបានភ្ជាប់នៅចុង 3′ នៃ primer ពីព្រោះការបញ្ចូលមូលដ្ឋាន 3 ចុងក្រោយនៅចុងបញ្ចប់ 3′ គឺគ្រប់គ្រាន់ដើម្បីចាប់ផ្តើម PCR នៅកន្លែងខុស។កំហាប់ primer ខ្ពស់ (1μM ទៅ 3μM) ត្រូវបានប្រើព្រោះ primers នៅក្នុងល្បាយឧបសម្ព័ន្ធជាច្រើនមិនជាក់លាក់ចំពោះគំរូគោលដៅ។
វត្ថុធាតុដើម PCRគ្រប់គ្រង
1. បរិមាណបឋម
កំហាប់នៃ primer នីមួយៗគឺ 0.1 ~ 1umol ឬ 10 ~ 100pmol ។វាជាការល្អប្រសើរជាងមុនដើម្បីផលិតលទ្ធផលដែលត្រូវការជាមួយនឹងបរិមាណទាបបំផុតនៃ primer ។កំហាប់ខ្ពស់នៃ primer នឹងបណ្តាលឱ្យមានការពង្រីកមិនស៊ីគ្នា និងមិនជាក់លាក់ និងបង្កើនឱកាសនៃការបង្កើត dimers រវាង primers ។
2. ការផ្តោតអារម្មណ៍បឋម
ការផ្តោតអារម្មណ៍នៃ primers ប៉ះពាល់ដល់ភាពជាក់លាក់។កំហាប់ primer ល្អបំផុតជាទូទៅគឺនៅចន្លោះ 0.1 និង 0.5μM។ការប្រមូលផ្តុំ primer ខ្ពស់នាំទៅដល់ការពង្រីកផលិតផលដែលមិនជាក់លាក់។
3. សីតុណ្ហភាព Annealing នៃ primer
ប៉ារ៉ាម៉ែត្រសំខាន់មួយទៀតសម្រាប់ primers គឺសីតុណ្ហភាពរលាយ (Tm) ។នេះគឺជាសីតុណ្ហភាពនៅពេលដែល 50% នៃ primers និងលំដាប់បន្ថែមត្រូវបានតំណាងថាជាម៉ូលេគុល DNA ពីរខ្សែ។Tm ត្រូវបានទាមទារដើម្បីកំណត់សីតុណ្ហភាពកំដៅ PCR ។តាមឧត្ដមគតិ សីតុណ្ហភាព annealing គឺទាបគ្រប់គ្រាន់ដើម្បីធានាបាននូវប្រសិទ្ធភាព annealing នៃ primers ជាមួយនឹងលំដាប់គោលដៅ ប៉ុន្តែខ្ពស់គ្រប់គ្រាន់ដើម្បីកាត់បន្ថយការចង nonspecific ។សីតុណ្ហភាព annealing សមហេតុផលពី 55 ℃ទៅ 70 ℃។សីតុណ្ហភាពនៃការដាក់ជាទូទៅត្រូវបានកំណត់ 5 ℃ទាបជាង Tm នៃ primer ។
មានរូបមន្តជាច្រើនសម្រាប់កំណត់ Tm ដែលប្រែប្រួលយ៉ាងខ្លាំងអាស្រ័យលើរូបមន្តដែលបានប្រើ និងលំដាប់នៃ primers ។ដោយសារតែរូបមន្តភាគច្រើនផ្តល់នូវតម្លៃ Tm ប៉ាន់ស្មាន សីតុណ្ហភាពនៃការស្រមុកទាំងអស់គ្រាន់តែជាចំណុចចាប់ផ្តើមប៉ុណ្ណោះ។ភាពជាក់លាក់អាចត្រូវបានធ្វើឱ្យប្រសើរឡើងដោយការវិភាគប្រតិកម្មជាច្រើនដែលបង្កើនសីតុណ្ហភាព annealing ជាលំដាប់។ចាប់ផ្តើមនៅក្រោម Tm-5 ℃ដែលបានប៉ាន់ប្រមាណហើយបង្កើនសីតុណ្ហភាព annealing បន្តិចម្តង ៗ ក្នុងការបង្កើន 2 ℃។សីតុណ្ហភាព annealing ខ្ពស់នឹងកាត់បន្ថយការបង្កើត primer dimers និងផលិតផលមិនជាក់លាក់។ដើម្បីទទួលបានលទ្ធផលល្អបំផុត ថ្នាំ primers ទាំងពីរគួរតែមានតម្លៃ Tm ប្រហាក់ប្រហែល។ប្រសិនបើភាពខុសគ្នានៃ Tm នៃគូ primer គឺច្រើនជាង 5 ℃ នោះ primers នឹងបង្ហាញពីការចាប់ផ្តើមមិនពិតដ៏សំខាន់ដោយប្រើសីតុណ្ហភាព annealing ទាបនៅក្នុងវដ្ត។ប្រសិនបើ primers Tm ទាំងពីរខុសគ្នា កំណត់សីតុណ្ហភាព annealing ដល់ 5 ℃ ទាបជាង Tm ទាបបំផុត។ជាជម្រើស ដើម្បីបង្កើនភាពជាក់លាក់ វដ្តចំនួន 5 អាចត្រូវបានអនុវត្តដំបូងនៅសីតុណ្ហភាព annealing ដែលបានរចនាឡើងសម្រាប់ Tm ខ្ពស់ជាង បន្តដោយវដ្តដែលនៅសល់នៅសីតុណ្ហភាព annealing ដែលត្រូវបានរចនាឡើងសម្រាប់ Tm ទាប។នេះអនុញ្ញាតឱ្យទទួលបានច្បាប់ចម្លងមួយផ្នែកនៃគំរូទិសដៅ ក្រោមលក្ខខណ្ឌតឹងតែង។
4. ភាពបរិសុទ្ធ និងស្ថេរភាពបឋម
ភាពបរិសុទ្ធស្តង់ដារនៃ primers ផ្ទាល់ខ្លួនគឺគ្រប់គ្រាន់សម្រាប់កម្មវិធី PCR ភាគច្រើន។ការដកក្រុម benzoyl និង isobutylyl ចេញដោយ desalting គឺតិចតួចបំផុត ដូច្នេះហើយមិនជ្រៀតជ្រែកជាមួយ PCR ទេ។កម្មវិធីមួយចំនួនទាមទារឱ្យមានការបន្សុត ដើម្បីដកចេញនូវលំដាប់ដែលមិនមានប្រវែងពេញលេញនៅក្នុងដំណើរការសំយោគ។លំដាប់កាត់បន្ថយទាំងនេះកើតឡើងដោយសារតែប្រសិទ្ធភាពនៃគីមីសាស្ត្រសំយោគ DNA មិន 100% ។នេះគឺជាដំណើរការរាងជារង្វង់ដែលប្រើប្រតិកម្មគីមីដដែលៗ ដោយសារមូលដ្ឋាននីមួយៗត្រូវបានបន្ថែមដើម្បីបង្កើត DNA ពី 3′ ទៅ 5′ ។អ្នកអាចបរាជ័យក្នុងវដ្តទាំងពីរ។ថ្នាំ primers វែង ជាពិសេស មូលដ្ឋានធំជាង 50 មានសមាមាត្រដ៏ធំនៃលំដាប់កាត់ ហើយអាចត្រូវការការបន្សុត។
ទិន្នផលនៃ primers ត្រូវបានរងផលប៉ះពាល់ដោយប្រសិទ្ធភាពនៃគីមីសាស្ត្រសំយោគនិងវិធីសាស្រ្តបន្សុត។ក្រុមហ៊ុនជីវឱសថ ដូចជា Cytology និង Shengong ទាំងអស់ប្រើឯកតា OD អប្បបរមា ដើម្បីធានាបាននូវទិន្នផលសរុបនៃ oligonucleoside ។primers ផ្ទាល់ខ្លួនត្រូវបានដឹកជញ្ជូនក្នុងទម្រង់ម្សៅស្ងួត។វាជាការល្អបំផុតក្នុងការរំលាយ primers ក្នុង TE ឡើងវិញដើម្បីឱ្យកំហាប់ចុងក្រោយគឺ 100μM។TE គឺល្អជាងទឹក deionized ព្រោះ pH នៃទឹកជាញឹកញាប់មានជាតិអាស៊ីត ហើយនឹងបណ្តាលឱ្យ hydrolysis នៃ oligonucleosides ។
ស្ថេរភាពនៃ primers អាស្រ័យលើលក្ខខណ្ឌផ្ទុក។ម្សៅស្ងួត និងថ្នាំ primers រលាយគួររក្សាទុកនៅ -20 ℃។ថ្នាំ primers រំលាយនៅក្នុង TE នៅកំហាប់ធំជាង 10μM អាចត្រូវបានរក្សាទុកយ៉ាងស្ថិតស្ថេរនៅ -20 ℃ សម្រាប់រយៈពេល 6 ខែ ប៉ុន្តែអាចរក្សាទុកបានតែនៅសីតុណ្ហភាពបន្ទប់ (15 ℃ ដល់ 30 ℃) ក្នុងរយៈពេលតិចជាង 1 សប្តាហ៍។ម្សៅទ្រនាប់ស្ងួតអាចរក្សាទុកនៅសីតុណ្ហភាព -20 C យ៉ាងហោចណាស់ 1 ឆ្នាំ និងនៅសីតុណ្ហភាពបន្ទប់ (15 C ដល់ 30 C) រហូតដល់ 2 ខែ។
5. អង់ស៊ីម និងការប្រមូលផ្តុំរបស់វា។
នាពេលបច្ចុប្បន្ននេះ Taq DNA polymerase ដែលត្រូវបានប្រើប្រាស់គឺជាមូលដ្ឋាននៃអង់ស៊ីមវិស្វកម្មហ្សែនដែលត្រូវបានសំយោគដោយបាក់តេរី coliform ។បរិមាណអង់ស៊ីមដែលត្រូវការសម្រាប់កាតាលីករប្រតិកម្ម PCR ធម្មតាគឺប្រហែល 2.5U (សំដៅទៅលើបរិមាណប្រតិកម្មសរុបនៃ 100ul) ។ប្រសិនបើការផ្តោតអារម្មណ៍គឺខ្ពស់ពេក, វាអាចនាំឱ្យមាន amplification មិនជាក់លាក់;ប្រសិនបើកំហាប់ទាបពេក បរិមាណផលិតផលសំយោគនឹងត្រូវបានកាត់បន្ថយ។
6. គុណភាពនិងការប្រមូលផ្តុំនៃ dNTP
គុណភាពនៃ dNTP គឺទាក់ទងយ៉ាងជិតស្និទ្ធទៅនឹងការប្រមូលផ្តុំ និងប្រសិទ្ធភាពនៃការពង្រីក PCR ។ម្សៅ dNTP មានលក្ខណៈជាក្រឡា ហើយភាពប្រែប្រួលរបស់វាបាត់បង់សកម្មភាពជីវសាស្រ្តប្រសិនបើវាត្រូវបានរក្សាទុកមិនត្រឹមត្រូវ។ដំណោះស្រាយ dNTP មានជាតិអាស៊ីត ហើយគួរប្រើក្នុងកំហាប់ខ្ពស់ ជាមួយនឹងដំណោះស្រាយសតិបណ្ដោះអាសន្ន 1M NaOH ឬ 1M Tris.HCL ដើម្បីកែតម្រូវ PH របស់វាទៅ 7.0 ~ 7.5 ចំនួនតូចនៃការវេចខ្ចប់រង ការផ្ទុកកកនៅ -20 ℃។ការរលាយកកច្រើននឹងធ្វើឱ្យ dNTP ថយចុះ។នៅក្នុងប្រតិកម្ម PCR dNTP គួរតែមាន 50 ~ 200umol / L ។ជាពិសេសការយកចិត្តទុកដាក់គួរតែត្រូវបានបង់ទៅការផ្តោតអារម្មណ៍នៃ DNTPS ទាំងបួនគួរតែស្មើគ្នា (ការរៀបចំ mole ស្មើគ្នា) ។ប្រសិនបើការផ្តោតអារម្មណ៍នៃមួយក្នុងចំណោមពួកគេខុសពីអ្នកដទៃ (ខ្ពស់ជាងឬទាបជាង) ភាពមិនស៊ីសង្វាក់គ្នានឹងត្រូវបានបង្កឡើង។ការផ្តោតអារម្មណ៍ទាបពេកនឹងកាត់បន្ថយទិន្នផលនៃផលិតផល PCR ។dNTP អាចផ្សំជាមួយ Mg2+ និងកាត់បន្ថយកំហាប់នៃ Mg2+ ដោយឥតគិតថ្លៃ។
7. គំរូ (ហ្សែនគោលដៅ) អាស៊ីត nucleic
បរិមាណ និងកម្រិតបន្សុតនៃអាស៊ីត nucleic គំរូគឺជាតំណភ្ជាប់ដ៏សំខាន់មួយសម្រាប់ភាពជោគជ័យ ឬបរាជ័យនៃ PCR ។វិធីសាស្រ្តបន្សុត DNA ប្រពៃណីជាធម្មតាប្រើ SDS និង protease K ដើម្បីរំលាយ និងចោលសំណាក។មុខងារចម្បងរបស់ SDS គឺ៖ រំលាយជាតិខ្លាញ់ និងប្រូតេអ៊ីននៅលើភ្នាសកោសិកា ដូច្នេះបំផ្លាញភ្នាសកោសិកាដោយការរំលាយប្រូតេអ៊ីនភ្នាស និងបំបែកប្រូតេអ៊ីននុយក្លេអ៊ែរនៅក្នុងកោសិកា SDS ក៏អាចផ្សំជាមួយប្រូតេអ៊ីន និង precipitate ផងដែរ។Protease K អាច hydrolyze និងរំលាយប្រូតេអ៊ីន ជាពិសេសអ៊ីស្តូនដែលចងភ្ជាប់ជាមួយ DNA ហើយបន្ទាប់មកប្រើសារធាតុរំលាយសរីរាង្គ phenol និង chloroform ដើម្បីទាញយកប្រូតេអ៊ីន និងសមាសធាតុកោសិកាផ្សេងទៀត ហើយប្រើអេតាណុល ឬអាល់កុល isopropyl ដើម្បី precipitate អាស៊ីត nucleic ។អាស៊ីតនុយក្លេអ៊ីកដែលបានស្រង់ចេញអាចត្រូវបានប្រើជាគំរូសម្រាប់ប្រតិកម្ម PCR ។សម្រាប់សំណាកការរកឃើញគ្លីនិកទូទៅ វិធីសាស្ត្ររហ័ស និងសាមញ្ញអាចប្រើដើម្បីរំលាយកោសិកា lysate ភ្នាក់ងារបង្កជំងឺ រំលាយ និងយកប្រូតេអ៊ីនចេញពីក្រូម៉ូសូមទៅជាហ្សែនគោលដៅដោយឥតគិតថ្លៃ ហើយប្រើដោយផ្ទាល់សម្រាប់ការពង្រីក PCR ។ការទាញយកគំរូ RNA ជាធម្មតាប្រើវិធីសាស្ត្រ guanidine isothiocyanate ឬ protease K ដើម្បីការពារ RNase ពីការបំផ្លាញ RNA ។
កំហាប់ 8.Mg2+
Mg2+ មានឥទ្ធិពលយ៉ាងសំខាន់ទៅលើភាពជាក់លាក់ និងទិន្នផលនៃការពង្រីក PCR ។នៅក្នុងប្រតិកម្ម PCR ជាទូទៅនៅពេលដែលកំហាប់នៃ dNTP ផ្សេងៗគឺ 200umol/L កំហាប់សមស្របនៃ Mg2+ គឺ 1.5 ~ 2.0mmol/L ។កំហាប់ Mg2+ ខ្ពស់ពេក ភាពជាក់លាក់នៃប្រតិកម្មថយចុះ ការពង្រីកមិនជាក់លាក់កើតឡើង កំហាប់ទាបពេកនឹងកាត់បន្ថយសកម្មភាពរបស់ Taq DNA polymerase ដែលជាលទ្ធផលកាត់បន្ថយផលិតផលប្រតិកម្ម។
អ៊ីយ៉ុងម៉ាញ៉េស្យូមប៉ះពាល់ដល់ទិដ្ឋភាពជាច្រើននៃ PCR ដូចជាសកម្មភាព DNA polymerase ដែលប៉ះពាល់ដល់ទិន្នផល។ឧទាហរណ៍មួយទៀតគឺ primer annealing ដែលប៉ះពាល់ដល់ភាពជាក់លាក់។dNTP និងគំរូភ្ជាប់ទៅនឹងអ៊ីយ៉ុងម៉ាញេស្យូមដោយកាត់បន្ថយបរិមាណនៃអ៊ីយ៉ុងម៉ាញេស្យូមដោយឥតគិតថ្លៃដែលត្រូវការសម្រាប់សកម្មភាពអង់ស៊ីម។កំហាប់អ៊ីយ៉ុងម៉ាញេស្យូមល្អបំផុតប្រែប្រួលសម្រាប់គូ primer និងគំរូផ្សេងៗគ្នា ប៉ុន្តែកំហាប់ចាប់ផ្តើម PCR ធម្មតាជាមួយ 200μM dNTP គឺ 1.5mM (ចំណាំ៖ សម្រាប់ PCR បរិមាណជាក់ស្តែង សូមប្រើដំណោះស្រាយម៉ាញ៉េស្យូមអ៊ីយ៉ុងពី 3 ទៅ 5mM ជាមួយនឹងការស៊ើបអង្កេត fluorescent) ។កំហាប់ខ្ពស់នៃអ៊ីយ៉ុងម៉ាញេស្យូមឥតគិតថ្លៃបង្កើនទិន្នផល ប៉ុន្តែក៏បង្កើនការពង្រីកដែលមិនជាក់លាក់ និងកាត់បន្ថយភាពស្មោះត្រង់ផងដែរ។ដើម្បីកំណត់កំហាប់ល្អបំផុត ការធ្វើតេត្រាអ៊ីយ៉ុងម៉ាញេស្យូមត្រូវបានអនុវត្តក្នុងការកើនឡើង 0.5mM ពី 1mM ទៅ 3mM ។ដើម្បីកាត់បន្ថយការពឹងផ្អែកលើការបង្កើនប្រសិទ្ធភាពអ៊ីយ៉ុងម៉ាញេស្យូម Platinum Taq DNA polymerase អាចត្រូវបានប្រើ។Platinum Taq DNA polymerase អាចរក្សាមុខងារលើជួរដ៏ធំទូលាយនៃកំហាប់អ៊ីយ៉ុងម៉ាញេស្យូមជាង Taq DNA polymerase ហើយដូច្នេះទាមទារការបង្កើនប្រសិទ្ធភាពតិច។
9. Pcr-ផ្សព្វផ្សាយសារធាតុបន្ថែម
ការបង្កើនប្រសិទ្ធភាពនៃសីតុណ្ហភាព annealing ការរចនា primer និងកំហាប់អ៊ីយ៉ុងម៉ាញ៉េស្យូមគឺគ្រប់គ្រាន់សម្រាប់ការពង្រីកជាក់លាក់ខ្ពស់នៃគំរូភាគច្រើន។ទោះយ៉ាងណាក៏ដោយ គំរូមួយចំនួន រួមទាំងអ្នកដែលមានមាតិកា GC ខ្ពស់ ទាមទារវិធានការបន្ថែម។សារធាតុបន្ថែមដែលប៉ះពាល់ដល់សីតុណ្ហភាពរលាយនៃ DNA ផ្តល់នូវវិធីមួយផ្សេងទៀតដើម្បីកែលម្អភាពជាក់លាក់នៃផលិតផល និងទិន្នផល។តម្រូវឱ្យមានការបង្ហាញទម្រង់ពេញលេញសម្រាប់លទ្ធផលល្អបំផុត។
លើសពីនេះទៀតរចនាសម្ព័ន្ធបន្ទាប់បន្សំការពារការចង primer និងផ្នែកបន្ថែមអង់ស៊ីម។
សារធាតុបន្ថែម PCR រួមមាន formamide, DMSO, glycerin, betaine និង PCRx Enhancer Solution បង្កើនការពង្រីក។យន្តការដែលអាចកើតមានរបស់ពួកគេគឺកាត់បន្ថយសីតុណ្ហភាពរលាយ ដូច្នេះវាជួយដល់ការបិទភ្ជាប់នៃ primers និងជួយផ្នែកបន្ថែម DNA polymerase តាមរយៈតំបន់រចនាសម្ព័ន្ធបន្ទាប់បន្សំ។PCRx Solution មានអត្ថប្រយោជន៍ផ្សេងទៀត។ការបង្កើនប្រសិទ្ធភាពអ៊ីយ៉ុងម៉ាញេស្យូមតិចតួចគឺត្រូវបានទាមទារនៅពេលប្រើជាមួយ Platinum Taq DNA polymerase និង Platinum Pfx DNA polymerase ។ដូច្នេះបច្ចេកទេសផ្លាទីនត្រូវបានផ្សំជាមួយនឹងការបន្ថែមដើម្បីបង្កើនភាពជាក់លាក់ខណៈពេលដែលកាត់បន្ថយការពឹងផ្អែកនៃវិធីសាស្រ្តទីបីគឺការបង្កើនប្រសិទ្ធភាពអ៊ីយ៉ុងម៉ាញ៉េស្យូម។ដើម្បីទទួលបានលទ្ធផលល្អបំផុត ការប្រមូលផ្តុំសារធាតុបន្ថែមគួរតែត្រូវបានធ្វើឱ្យប្រសើរ ជាពិសេស DMSO, formamide និង glycerol ដែលរារាំង Taq DNA polymerase ។

10. ការចាប់ផ្តើមក្តៅ
ការចាប់ផ្តើមក្តៅ PCR គឺជាវិធីសាស្រ្តដ៏សំខាន់បំផុតមួយដើម្បីកែលម្អភាពជាក់លាក់របស់ PCR បន្ថែមលើការរចនា primer ដ៏ល្អ។ទោះបីជាសីតុណ្ហភាពពន្លូតល្អបំផុតនៃ Taq DNA polymerase គឺ 72 ℃ក៏ដោយ សារធាតុ polymerase នៅតែសកម្មនៅសីតុណ្ហភាពបន្ទប់។ដូច្នេះផលិតផលមិនជាក់លាក់ត្រូវបានផលិតនៅពេលដែលសីតុណ្ហភាពកាន់ទាបជាងសីតុណ្ហភាព annealing កំឡុងពេលរៀបចំប្រតិកម្ម PCR និងនៅដើមដំបូងនៃវដ្តកម្ដៅ។នៅពេលដែលបង្កើតឡើង ផលិតផលដែលមិនជាក់លាក់ទាំងនេះត្រូវបានពង្រីកយ៉ាងមានប្រសិទ្ធភាព។Hot-start PCR មានប្រសិទ្ធភាពជាពិសេសនៅពេលដែលគេហទំព័រដែលប្រើសម្រាប់ការរចនាបឋមត្រូវបានកំណត់ដោយទីតាំងនៃធាតុហ្សែន ដូចជាការផ្លាស់ប្តូរតាមគេហទំព័រ ការក្លូនការបញ្ចេញមតិ ឬការសាងសង់ និងការរៀបចំធាតុហ្សែនដែលប្រើសម្រាប់វិស្វកម្ម DNA ។
វិធីសាស្រ្តទូទៅមួយដើម្បីកំណត់សកម្មភាពរបស់ Taq DNA polymerase គឺដើម្បីរៀបចំដំណោះស្រាយប្រតិកម្ម PCR នៅលើទឹកកក ហើយដាក់វានៅក្នុងឧបករណ៍ PCR ដែលកំដៅមុន។វិធីសាស្រ្តនេះគឺសាមញ្ញនិងមានតំលៃថោកប៉ុន្តែវាមិនបំពេញសកម្មភាពនៃអង់ស៊ីមទេហើយដូច្នេះមិនលុបបំបាត់ទាំងស្រុងនូវការពង្រីកនៃផលិតផលដែលមិនជាក់លាក់នោះទេ។
ការដាក់កំដៅ ពន្យារការសំយោគ DNA ដោយរារាំងសមាសធាតុសំខាន់មួយ រហូតដល់បរិធាន PCR ឈានដល់សីតុណ្ហភាព denaturation ។វិធីសាស្រ្តចាប់ផ្តើមកម្ដៅដោយដៃភាគច្រើន រួមទាំងការពន្យាពេលបន្ថែមនៃ Taq DNA polymerase គឺមានភាពលំបាក ជាពិសេសសម្រាប់កម្មវិធីដែលមានថាមពលខ្ពស់។វិធីសាស្ត្រដាក់កំដៅផ្សេងទៀត ប្រើប្រឡោះក្រមួន ដើម្បីរុំព័ទ្ធសមាសធាតុសំខាន់ៗ រួមទាំងអ៊ីយ៉ុងម៉ាញេស្យូម ឬអង់ស៊ីម ឬដើម្បីបំបែកសមាសធាតុប្រតិកម្មដូចជា គំរូ និងសតិបណ្ដោះអាសន្ន។ក្នុងអំឡុងពេលនៃវដ្តកំដៅ សមាសធាតុផ្សេងៗត្រូវបានបញ្ចេញ និងលាយបញ្ចូលគ្នានៅពេលដែលក្រមួនរលាយ។ដូចវិធីសាស្រ្តនៃការចាប់ផ្តើមក្តៅដោយដៃ វិធីសាស្ត្រការពារក្រមួនគឺពិបាក និងងាយនឹងមានការចម្លងរោគ និងមិនស័ក្តិសមសម្រាប់កម្មវិធីដែលមានចរន្តខ្ពស់នោះទេ។
Platinum DNA polymerase មានភាពងាយស្រួល និងមានប្រសិទ្ធភាពសម្រាប់ PCR ចាប់ផ្តើមក្តៅដោយស្វ័យប្រវត្តិ។Platinum Taq DNA polymerase មានផ្ទុកនូវសារធាតុ Taq DNA polymerase ដែលផ្សំគ្នាជាមួយនឹងអង្គបដិប្រាណ monoclonal ប្រឆាំងនឹង Taq DNA polymerase ។អង្គបដិប្រាណត្រូវបានបង្កើតឡើងដោយ PCR ដើម្បីរារាំងសកម្មភាពអង់ស៊ីមក្នុងអំឡុងពេលរក្សាសីតុណ្ហភាពយូរ។Taq DNA polymerase ត្រូវបានបញ្ចេញទៅក្នុងប្រតិកម្មកំឡុងពេលអ៊ីសូឡង់ 94 ℃ នៃជំហាន denaturation ដោយស្ដារឡើងវិញនូវសកម្មភាព polymerase ពេញលេញ។ផ្ទុយទៅនឹងសារធាតុ Taq DNA polymerase ដែលបានកែប្រែគីមីសម្រាប់ការចាប់ផ្តើមកម្ដៅ អង់ស៊ីមផ្លាទីនមិនតម្រូវឱ្យមានអ៊ីសូឡង់យូរនៅសីតុណ្ហភាព 94 ℃ (10 ទៅ 15 នាទី) ដើម្បីធ្វើឱ្យប៉ូលីមេរ៉ាសសកម្មនោះទេ។ជាមួយនឹង PlatinumTaq DNA polymerase សកម្មភាព 90% នៃ Taq DNA polymerase ត្រូវបានស្ដារឡើងវិញបន្ទាប់ពី 2 នាទីនៅ 94 ℃។

11. Nest-PCR
ការពង្រីកជាបន្តបន្ទាប់ដោយប្រើប្រាស់ថ្នាំ primers ដែលភ្ជាប់មកជាមួយអាចធ្វើឱ្យប្រសើរឡើងនូវភាពជាក់លាក់ និងភាពប្រែប្រួល។ជុំទីមួយគឺជាការពង្រីកស្តង់ដារពី 15 ទៅ 20 វដ្ត។ប្រភាគតូចមួយនៃផលិតផល amplification ដំបូងត្រូវបានពនឺពី 100 ទៅ 1000 ដង ហើយបន្ថែមទៅជុំទីពីរនៃ amplification សម្រាប់ 15 ទៅ 20 វដ្ត។ជាជម្រើស ផលិតផលពង្រីកដំបូងអាចមានទំហំដោយការបន្សុតដោយជែល។primer nested ត្រូវបានប្រើនៅក្នុងជុំទីពីរនៃ amplification ដែលអាចភ្ជាប់ទៅនឹងលំដាប់គោលដៅនៅខាងក្នុង primer ទីមួយ។ការប្រើប្រាស់ PCR ដែលត្រូវបានភ្ជាប់គ្នាកាត់បន្ថយលទ្ធភាពនៃការពង្រីកគេហទំព័រគោលដៅច្រើន ពីព្រោះមានលំដាប់គោលដៅតិចតួចដែលបំពេញបន្ថែមទៅនឹងសំណុំនៃ primers ទាំងពីរ។ចំនួនវដ្តសរុបដូចគ្នា (ពី 30 ទៅ 40) ជាមួយនឹង primers ដូចគ្នាបានពង្រីកតំបន់ដែលមិនជាក់លាក់។Nested PCR បង្កើនភាពប្រែប្រួលនៃលំដាប់គោលដៅមានកំណត់ (ឧទាហរណ៍ mrnas កម្រ) និងធ្វើអោយប្រសើរឡើងនូវភាពជាក់លាក់នៃ PCRS ពិបាក (ឧទាហរណ៍ 5′ RACE)។
12. PCR ធ្លាក់ចុះ
Descending PCR ធ្វើអោយប្រសើរឡើងនូវភាពជាក់លាក់ដោយប្រើលក្ខខណ្ឌតឹងណែនសម្រាប់វដ្តពីរបីដំបូងនៃ PCR ។វដ្តចាប់ផ្តើមនៅសីតុណ្ហភាព annealing ប្រហែល 5 ℃ខ្ពស់ជាង Tm ប៉ាន់ស្មានបន្ទាប់មកវដ្តនីមួយៗត្រូវបានកាត់បន្ថយដោយ 1 ℃ទៅ 2 ℃រហូតដល់សីតុណ្ហភាព annealing ទាបជាង Tm 5 ℃។មានតែគំរូទិសដៅដែលមានភាពដូចគ្នាខ្ពស់បំផុតប៉ុណ្ណោះនឹងត្រូវបានពង្រីក។ផលិតផលទាំងនេះបន្តពង្រីកនៅក្នុងវដ្តជាបន្តបន្ទាប់ ដោយប្រមូលផ្តុំផលិតផលដែលមិនជាក់លាក់។Descending PCR មានប្រយោជន៍សម្រាប់វិធីសាស្រ្តដែលកម្រិតនៃភាពដូចគ្នារវាង primer និងគំរូគោលដៅមិនត្រូវបានគេស្គាល់ ដូចជា AFLP DNA fingerprinting។
ឧបករណ៍ PCR ពាក់ព័ន្ធ
វីរបុរស 2 × PCRTMប្រព័ន្ធលាយមានភាពអត់ធ្មត់ខ្ពស់ចំពោះ PCR inhibitors ជាងប្រព័ន្ធ PCR Mix ធម្មតា ហើយអាចដោះស្រាយយ៉ាងងាយស្រួលជាមួយនឹងការពង្រីក PCR នៃគំរូស្មុគស្មាញផ្សេងៗ។ប្រព័ន្ធប្រតិកម្មតែមួយគត់ និងប្រសិទ្ធភាពខ្ពស់ Taq Hero ធ្វើឱ្យប្រតិកម្ម PCR មានប្រសិទ្ធភាព ភាពជាក់លាក់ និងភាពប្រែប្រួលខ្ពស់។
ប្រសិទ្ធភាពពង្រីកកាន់តែខ្ពស់។
វាមានសកម្មភាព 5'→3' DNA polymerase និង 5'→3' សកម្មភាព exonuclease ដោយគ្មានសកម្មភាព 3'→5' exonuclease ។
ពេលវេលាពិត PCR Easyᵀᴹ-SYBR Green I Kit
ជាក់លាក់ - សតិបណ្ដោះអាសន្នដែលបានកែលម្អ និងអង់ស៊ីម Taq ចាប់ផ្តើមក្តៅអាចការពារការពង្រីកមិនជាក់លាក់ និងការបង្កើត primer dimer
ភាពប្រែប្រួលខ្ពស់ - អាចរកឃើញច្បាប់ចម្លងទាបនៃគំរូ
RT-PCR ងាយស្រួលᵀᴹ I (ជំហានមួយ)
កញ្ចប់នេះប្រើសារធាតុចម្លងបញ្ច្រាស Foregene តែមួយគត់ និង Foregene HotStar Taq DNA Polymerase រួមបញ្ចូលគ្នាជាមួយប្រព័ន្ធប្រតិកម្មពិសេសមួយ ដើម្បីបង្កើនប្រសិទ្ធភាពនៃការពង្រីក និងភាពជាក់លាក់នៃប្រតិកម្ម។
ពេលវេលាបង្ហោះ៖ ឧសភា-០៩-២០២៣



